有機化学の基本となる反応の一つである酸化反応は,実験室レベルの合成からファインケミカルズ(試薬・医薬品など)の合成,さらに石油化学工業に至るまで幅広く利用されている。その中でも炭化水素類の水酸化は,有機合成において重要な反応である。炭化水素類を水酸化する方法として,まず原料の炭化水素類を目的生成物に対応するケトンやアルデヒドあるいはカルボン酸に酸化し,それを適切な還元処理を行うことで目的のアルコールを得る方法が挙げられる。この方法を用いる理由として,炭化水素の酸化反応を行う際,アルコールが一旦生成するが,そのアルコールが優先的に再酸化されてしまうことが挙げられる。近年提唱されている「グリーンサステイナブルケミストリー」の概念1)からすれば,反応が多段階になればなるほど,廃液,副生物の量や種類が増え,環境負荷が大きくなるので,反応段数を減らすことが望ましい。古くから炭化水素類を一段階で直接水酸化してアルコールを生成する反応は高難度酸化反応の一つとされているが,一段階に短縮できれば,余分な反応試薬や有機溶媒などが削減でき,エネルギーの低減にも繋がり,環境負荷を低減できるため,現在でも特に注目されている。また,酸化反応は反応の制御といった観点から多くの問題を抱えている。例えば,種々の酸化反応には未だに量論量以上必要な酸化剤(クロム,マンガンなどを含む重金属塩,次亜塩素塩,硝酸など)が用いられており,それらを用いることで毒性重金属塩の処理などの問題も生じる。従って,水のみが副生成物として生じる酸化剤である分子状酸素や過酸化水素を用いて効率的かつ触媒的に有機基質の選択酸化反応を進行させることは大幅な汚染物質の低減につながる。また,反応溶媒を有機溶媒から水溶媒へシフトできると余分な有機物質を削減できるため,より環境に優しい反応系が構築できる。
酸化反応などで用いられている均一系触媒は,使用後の分離・回収,再利用が困難であり,また,触媒同士が反応して失活するなどの問題がある。そこで,触媒,触媒担体,吸着剤,物質分離材などに利用されているゼオライトやメソ多孔質シリカ2,3)に錯体を固定化して,上記の問題を解決する方法が検討されている。ゼオライトは,ソーダライトケージ,スーパーケージをはじめとする規定されたミクロ分子環境場を提供し,分子反応器として前述のような様々な機能を発揮する。本稿では,金属錯体内包触媒の最近の研究を概観して,著者らが行っている鉄錯体及び銅錯体をゼオライト細孔に内包した触媒を用いた有機基質に対する選択酸化反応について紹介する。
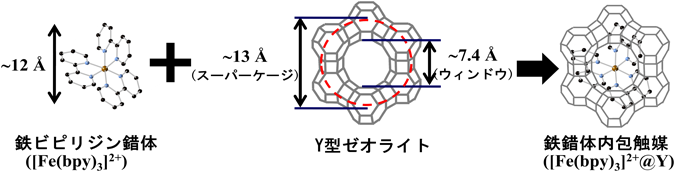
Y型ゼオライトのスーパーケージの内径は約13 Åであり,その最大入口直径は約7.4 Åである(図1)。Y型ゼオライトのスーパーケージ内に合成された嵩高い金属錯体(例えば,[Fe(bpy)3]2+錯体(直径約12 Å))はY型ゼオライトの最大入口直径よりも大きいため,スーパーケージから出ることができない4–6)。それゆえ,不均一系触媒として様々な用途で用いられている6–30)。これまでに報告されているゼオライトに内包させた金属錯体の一例を表1にまとめている。
表1 Y型ゼオライトに内包された遷移金属錯体と触媒反応への応用例| 内包された金属錯体 | 触媒反応への応用例 | 文献 |
| cis-[Mn(bpy)2]2+, a) | アルケンの選択的エポキシ化反応 | 7 |
| FePcb), Fe(TMP)c) | シクロヘキセン,シクロヘキサン,オクタンの酸化 | 8 |
| [M(SFCH)·xH2O] (M=Mn, Fe, Co, Ni, Cu)d) | シクロヘキサン酸化 | 9 |
| [VO(hap-dahp)]e) | スチレン,スルフィド類の酸化 | 10 |
| [Ru(salophen)]f) | アルケンの選択的エポキシ化反応 | 11 |
| [V(picolinic acid)2] | ベンゼン酸化 | 12 |
| [M(salen)] (M=Zn, Co, Mn, Fe, Cr)g) | シクロヘキセン,キシレンの酸化 | 13 |
| [M(ImzlH)]h), [M(PhBzlH)]i) (M=Ru, Cu, Zn) | エチルベンゼン,フェノール等の酸化 | 14,15 |
| [M(TMePP)] (M=Mn, Fe, Co)j) | リモネン酸化 | 16 |
| [M(L)] (M=Co, Ni, Cu)k) | ベンジルアルコール酸化 | 17 |
| [M{Me4(Et)2[14]tetraneN4}] (M=Cu, V, Co) | スチレン酸化 | 18 |
| [Mn2(HL)2(H2O)4]l) | シクロヘキセンのエポキシ化反応 | 19 |
| [Fe(phen)]2+, m) | メチルグリーンの光分解反応 | 20 |
| [M(bpy)3]2+ (M=Fe, Ru)a) | ベンゼン酸化,光を利用した選択的部分酸化反応 | 6,21–23 |
| [VO(L)]n) | アルデヒド類の還元反応 | 24 |
| [Cu(L1)]2+, [Cu(L2)]2+, o) | Henry反応 | 25 |
| [Cu(N2S2)]+, p) | プロパルギルアミン類の合成 | 26 |
| [PdCl2(phen)]m) | アミノカルボニル化反応 | 27 |
| [Ni(salen)]g) | メタノールの電極触媒的酸化反応 | 28,30 |
| [Ni{salnptn(4-OH)2}]q) | 糖類のサイズ選択的電極触媒反応 | 29 |
| a) bpy: 2,2'-bipyridine. b) Pc: phthalocyanine. c) TMP: tetramethylporphyrin. d) H2SFCH: (E)-N'-(2-hydroxybenzylidene)furan-2-carbohydrazide. e) hap-dahp: schiff base ligand derived from 2-hydroxyacetohenone and 1,3-diamino-2-hydroxypropane. f) salophen: (4-acetamidophenyl)2-hydroxybenzoate. g) salen: schiff base ligand derived from salicylaldehyde and 1,2-ethylenediamine. h) ImzlH: imidazole. i) PhBzlH: 2-phenylbenzimidazole. j) TMePP: 5,10,15,20-tetra(4-methoxyphenyl)porphyrin. k) H2 L: 2-[(2-hydroxy-phenylimino)-methyl]-4-(4-chloro-3-nitro-phenylazo)-phenol. l) HL: schiff base ligand derived from salicylaldehyde and L-tyrosine. m) phen: orthophenantroline. n) L: schiff base ligand derived from 2,4-hydroxyacetophenone and 2,2'-dimethylpropanediamine. o) L1: (1S, 2S)-N1,N2-bis(3-chlorobenzyl)cyclohexane-1,2-diamine, L2: (1S, 2S)-N1,N2-bis(4-chlorobenzyl)cyclohexane-1,2- diamine. p) N2S2: bis[2-(phenylthio)benzyldene]1,2-ethylenediamine. q) H2{salnptn(4-OH)2}: N,N'-bis(2,4-dihydroxyacetophenone)-2,2-dimethyl propandiimine. |
1994年,Jacobsら7)はMn-ビピリジン錯体を内包したゼオライト触媒を用いてアルケンの酸化反応を行い,選択的エポキシ化反応が進行することを報告した。それ以降,様々な遷移金属(Mn,Fe,Co,Ni,Cu,Zn,Ruなど)と配位子(シッフ塩基系(サレンなど),マクロサイクル系(フタロシアン,ポルフィリンなど),ピリジン・イミダゾール系多座配位子,三脚型配位子など)を用いた種々の遷移金属錯体をゼオライト細孔に導入し,過酸化水素などを酸化剤として用いた有機基質(フェノール,シクロヘキセン,スルフィド類など)に対する酸化反応が,数多くの研究者により報告され続けている6,8–19)。Fe錯体を導入したゼオライト触媒はメチルグリーンなどの有機色素の光分解反応に利用されており20),近年ではFeやRu錯体を内包した触媒を用い,光を利用した有機基質に対する選択的部分酸化反応などへ応用されている21–23)。
また,酸化反応だけでなく,V錯体を内包した触媒を用いたアルデヒドの還元反応や24),CuやPd錯体を内包した触媒を用いたHenry反応25),プロパギルアミン類の合成26),アミノカルボニル化反応27)への応用も試みられている。さらに,Ni錯体を内包した触媒を導入した電極を用いた電極触媒への応用事例も報告されており28–30),不均一系触媒としての用途はさらに拡大している。
3. 金属錯体内包ゼオライト触媒のキャラクタリゼーション
金属錯体内包ゼオライト触媒は,まず,錯体の中心金属をゼオライトにイオン交換した後,配位子を導入することで調製されている。例えば,鉄錯体内包ゼオライト触媒([Fe(bpy)3]2+@Y)は,Feイオン交換したY型ゼオライト(Fe-Y)に2,2'-ビピリジン(bpy)を添加して還流操作を行い,ソックスレー抽出を行うことで得られている31–34)。他にも予め遷移金属錯体を合成し,その錯体の周りにゼオライトを合成する方法(ゼオライト合成法)が報告されているが35,36),一般的ではない。
調製した試料は複数の分析手法を総合して目的の化合物であると同定される。[Fe(bpy)3]2+@Yでは,下記の(1)–(5)の実験結果を総合して,ゼオライト空孔内で[Fe(bpy)3]2+錯体が生成していると推定している31–34)。(1)元素分析(ICP, CHN)の結果より,Fe : bpy=1 : 3である。(2)FT-IRスペクトルにおいて,[Fe(bpy)]2+錯体由来のピークが確認できる。(3)Y型ゼオライトの(220)面(2θ=10°)と(311)面(12°)に帰属されるXRDピークの強度比がbpy添加前後で変化している4–6)(図2)。(4)TG/DTA曲線において,[Fe(bpy)3]2+@Y中のbpy配位子の燃焼開始温度が,ゼオライト外表面に存在する[Fe(bpy)3]2+錯体([Fe(bpy)3]2+/Na-Y)中のbpy配位子の燃焼開始温度よりも高い。これは[Fe(bpy)3]2+錯体がゼオライト骨格に守られているため,熱的安定性が向上するためである。(5)拡散反射UV-vis.スペクトル測定から(図3),[Fe(bpy)3]2+@Yでは290と531 nmにbpy由来のπ–π*遷移とMLCTに帰属可能な吸収がそれぞれ観察され,これらの吸収強度はFeのモル数を揃えた[Fe(bpy)3](ClO4)2+Na-Yのそれと比較して大きい。この[Fe(bpy)3]2+@Yでの大きな吸収強度は,ゼオライト細孔内に[Fe(bpy)3]2+錯体が分散していることに起因する。
さらに,金属錯体の立体構造や歪みを調べるために,ESRやXAFSも有効なツールである。[Cu(terpy)]2+@YのESRから,内包触媒中の銅イオン周りの配位構造が平面四角形型構造(g//=2.24, g⊥=2.05, |A//|=180 G)であることが示唆されている。また,[Cu(phthalocyanine)]2+@Yでは,ESRおよび計算から金属錯体が歪んでいることが明らかにされている37)。さらに,[Fe(bpy)3]2+@YのEXAFSスペクトルでは,金属錯体が歪んでいることが証明されている32)。
4. 鉄錯体内包ゼオライト触媒を用いた環状炭化水素類の選択的酸化反応
シクロヘキセンの酸化反応では,用いる触媒や酸化剤によってさまざまな生成物が生成する。特に触媒として鉄錯体を用いた検討は詳細に行われており,活性酸素種と生成物との対応も明らかとなってきている38,39)。内包触媒の特徴を明らかにするために,著者らによって行われた鉄ビピリジン錯体内包ゼオライト触媒([Fe(bpy)3]2+@Y)と均一系触媒([Fe(bpy)3](ClO4)2)とのシクロヘキセンの酸化反応結果の比較を記述する。
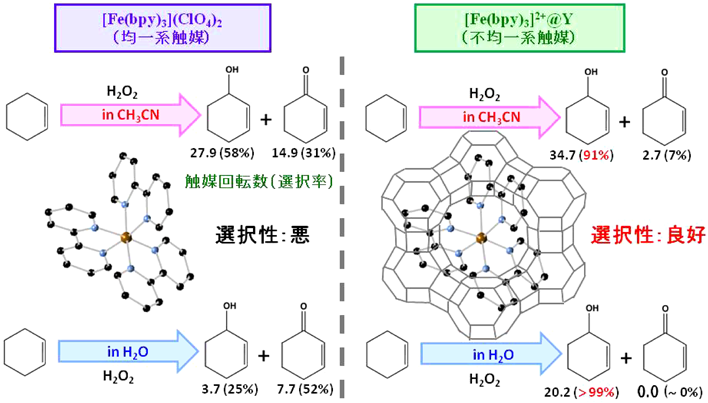
アセトニトリルと水を溶媒とし,[Fe(bpy)3]2+@Y内包触媒と[Fe(bpy)3](ClO4)2均一系触媒を用いた時のシクロヘキセンの酸化反応結果を図4に示す。アセトニトリル溶媒の場合,[Fe(bpy)3]2+@Y触媒の2-シクロヘキセン-1-オールの選択率は90%以上であり,[Fe(bpy)3](ClO4)2と大きく異なっていた。また,過酸化水素の有効利用率は,[Fe(bpy)3]2+@Yが[Fe(bpy)3](ClO4)2よりも高く,[Fe(bpy)3]2+@Yはシクロヘキセンの水酸化反応に対して有効な触媒である。さらに,[Fe(bpy)3]2+@Y触媒は,アセトニトリル中では少なくとも3回は活性,選択性を保ったまま繰り返し利用できることが証明されている。注目すべき点はこのシクロヘキサンの水酸化反応が水溶媒中でも進行することである。図4に示すように[Fe(bpy)3]2+@Yの方が[Fe(bpy)3](ClO4)2を用いたときよりも5倍以上多くの2-シクロヘキセン-1-オールを生成する。このように[Fe(bpy)3]2+@Yは水溶媒中においてもその触媒活性を維持し,ほぼ選択的に水酸化反応が進行する31–34)。
本触媒の反応機構についても検討されている31–34)。反応系中にラジカルトラップ剤(2,6-ジ-tert-ブチル4-メチルフェノール)を添加したところ,酸化生成物が観測されなかった。従って,[Fe(bpy)3]2+@Y触媒は過酸化水素と反応し,フェントン反応を経て,ラジカル活性種を形成して反応が進行していると考えられる。また,[Fe(bpy)3](ClO4)2を用いた2-シクロヘキセン-1-オールの酸化反応では,2-シクロヘキセン-1-オンが生成したことから,[Fe(bpy)3](ClO4)2触媒でのシクロヘキセンの酸化はオンからオールへ逐次的に反応が進行する(図5(ⅰ))。[Fe(bpy)3]2+@Yで高いアルコール選択性を示す理由として,Y型ゼオライト内に存在しているNa+イオンが2-シクロヘキセン-1-オールを捕捉するため,ケトンを生成する再酸化反応を抑制していることが考えられる(図5(ⅱ))。アルコールを捕捉できない(CH3)4N+イオンにNa+イオンの一部を交換した触媒を用いるとアルコール選択性が低下したことからも支持される。以上の結果から,[Fe(bpy)3]2+部位は触媒活性部位として酸化反応に寄与し,Na+イオン周りは親水性部位あるいは生成物捕捉部位として,アルコール選択性発現に寄与していると推測できる。また,[Fe(bpy)3]2+@Yでは,触媒内に親水性・疎水性の両方の部位ができ,その結果,水中でもシクロヘキセンの選択的水酸化反応を優先的に進行すると推察している。
[Fe(bpy)3]2+@Y触媒は過酸化水素を酸化剤として用いたベンゼンの酸化反応にも活性を示す40,41)。ベンゼンの酸化反応のアセトニトリル溶媒に対する水の添加を詳細に検討した結果(図6),水とアセトニトリルの体積比が1 : 1のときに,活性が飛躍的に向上した。水溶媒ではベンゼンの分散,アセトニトリル溶媒では過酸化水素の分散がそれぞれ触媒活性を低下させる問題点であり,混合溶媒となったことでそれぞれの溶媒の問題点が相殺されたため,活性が向上したと考えられている。また,均一系触媒[Fe(bpy)3](ClO4)2では,反応系中の過酸化水素はまだ残っていたにも関わらず,失活して5時間で反応がほぼ停止したのに対し,[Fe(bpy)3]2+@Yでは,反応速度はゆっくりであるが,生成物は増加し続け,24時間後の触媒回転数(TON)は,[Fe(bpy)3]2+@Y(TON=94)が[Fe(bpy)3](ClO4)2 (TON=83)を上回った。このように[Fe(bpy)3]2+@Yはベンゼン酸化に対しても良い触媒である40,41)。また,水溶媒中での[Fe(bpy)3]2+@Yのベンゼン酸化活性は低いが,ゼオライトのNa+イオンを他の金属カチオンに一部イオン交換すると活性が改善される。触媒活性の序列(Cs+>K+>Na+)は,水和イオン半径の序列(Cs+<K+<Na+)と概ね一致していることから,カチオンの水和イオン半径の違いにより[Fe(bpy)3]2+@Y中の空孔内の反応に有効な反応場(空隙)の広さが制限され,その反応場が広いほど触媒活性が向上すると考えられている40)。
上述したように鉄錯体内包ゼオライト触媒を利用すれば,過酸化水素による炭化水素の水酸化反応が触媒的に進行する。(1)環境に優しい水を溶媒とできること,(2)過酸化水素を酸化剤として用いることで反応後の副生成物が水のみであること,(3)反応温度が比較的低温(50°C)であること,(4)ゼオライト内に錯体を合成することで触媒の分離・回収が容易であること,(5)水酸化生成物を高い選択性で得られることなど多くの利点が挙げられる。
過酸化水素を用いた有機基質の部分酸化反応において,単核銅ハイドロパーオキサイド種は重要な活性種として考えられる。以前から水素結合相互作用や配位構造を制御した配位子を用いることにより室温で単離された単核銅ハイドロパーオキサイド種に関する様々な構造学的・分光学的情報が得られている42,43)。特に,平面四角形型構造を有する銅錯体は活性種の安定性が低下し,外部基質への酸化反応が進行することが明らかになっている42,44)。そこで,多孔質であるゼオライトの空孔内で平面四角形型構造を有する銅錯体を合成し,不均一系触媒として利用することが検討されている。
著者らはY型ゼオライトに[Cu(terpy)]2+錯体を固定化した触媒([Cu(terpy)]2+@Y)を設計・合成し,過酸化水素を酸化剤としたチオアニソールの酸化反応を行った。本反応ではスルホキサイドを選択的に生成し,スルホキサイドがさらに酸化されて生成するスルホンはほとんど観測されず,その触媒活性は均一系触媒である[Cu(terpy)(CH3CN)](ClO4)2と同等であった。また,[Cu(terpy)]2+@Yは少なくとも5回は触媒活性・スルホキサイド選択性を維持したまま再利用が可能である45)。
一般に,銅錯体と過酸化水素を用いたスルフィドの酸化反応機構は,銅錯体と過酸化水素の反応により酸化活性種(ここでは,Cu–OOH種)を生成する過程(Pathway A),酸化活性種によりスルフィドが酸化される過程(Pathway B)が考えられている。触媒・基質・酸化剤の物質量と反応速度を対数プロットしたところ,それぞれ直線関係が得られ,反応速度は触媒,基質,酸化剤の物質量に対して(1)式のように表せる。
この反応では,触媒(Cu)と酸化剤(H2O2)との反応,つまり,Cu–OOH種の生成反応が律速段階(Pathway A)であることが明らかである45)。[Cu(terpy)]2+@Yに過酸化水素を添加すると,380 nm付近にOOH−からCu2+へのLMCT遷移に帰属される吸収スペクトルが観測された。また,ESRパラメーター(g//=2.24, g⊥=2.06, |A//|=180 G)から,内包触媒中の銅イオン周りの配位構造が過酸化水素添加後も平面四角形構造を維持していることを確認された。以上の結果から,[Cu(terpy)]2+@Y中に平面四角形型構造を有する単核銅(Ⅱ)-ハイドロパーオキソ種が形成していることが考えられ,この活性種が室温でも安定に存在できることが明らかとなった45)。
[Cu (terpy)]2+@YとCu-Yを用いて,活性種の安定性および反応活性について検討されている。それぞれの銅含有触媒に過酸化水素を添加すると,[Cu(terpy)]2+@Yは黄緑色へ変化したが,Cu-Yは全く変化せず淡青色のままであった。過酸化水素を添加した後,遠心分離にて回収した触媒([[Cu(terpy)]2+@Y]*, [Cu-Y]*)を用いて,チオアニソールの酸化反応を行ったところ,[[Cu(terpy)]2+@Y]*では反応が徐々に進行し,スルホキサイドが選択的に生成した。触媒回転数(TON)は1で飽和し,銅に対して等量の活性種が生成していたことを示す結果が得られた。また,[Cu-Y]*では24時間後も全く反応が進行しなかった。このように,[Cu(terpy)]2+@Y中ではCu-Yとは異なり,活性なCu–OOH種が安定に存在できることが明らかにされている45)。
以上,金属錯体を内包したゼオライト触媒の有機基質に対する選択的酸化反応について我々の研究結果を中心に紹介した。前述したように金属錯体内包ゼオライト触媒の触媒設計のコンセプト自身は決して新しいものではないが,均一系触媒とは明らかに異なる新機能性が生れつつある。また,最近の分析機器の進歩で内包金属錯体の構造,反応性が徐々に明らかになってきている。我々の研究から,遷移金属錯体内包ゼオライト触媒の遷移金属錯体部分は触媒活性部位や疎水性部位として機能し,ナトリウムイオンなどのカチオン部位は生成物捕捉部位や親水性部位として機能する役割分担も明らかとなっている。現在,著者らの研究室では,触媒活性部位である遷移金属錯体部位や生成物捕捉部位であるカチオン部位について,遷移金属錯体内包ゼオライト触媒のファインチューニング,基質を種々のアルカン類や芳香族炭化水素類としたときの酸化反応系の開発も行っている。今後,金属錯体内包ゼオライト触媒の新しい触媒反応系が開発されることを期待したい。
謝辞Acknowledgments
本稿で紹介した我々の研究は,JSPS科研費25820394の助成を受けた。記してここに感謝の意を表します。
引用文献References
1) 決定版 感動する化学―未来をひらく化学の世界,日本化学会編,東京書籍,p. 8, 2010年.
2) C. Brémard, Coord. Chem. Rev., 178–180, 1647 (1998).
3) V. Ramamurthy, J. Photochem. Photobiol. C: Photochem. Rev., 1, 145 (2000).
4) Y. Umemura, Y. Minai, T. Tominaga, J. Phys. Chem. B, 103, 647 (1999).
5) R. Vijayalakshmi, S. K. Kulshreshtha, Microporous Mesporous Mater., 111, 449 (2008).
6) K. Mori, K. Kagohara, H. Yamashita, J. Phys. Chem. C, 112, 2593 (2008).
7) P.-P. Knops-Gerrits, D. D. Vos, F. Thibault-Starzyk, P. A. Jacobs, Nature, 369, 543 (1994).
8) F. Bedioui, Coord. Chem. Rev., 144, 39 (1995).
9) C. K. Modi, P. M. Trivedi, J. A. Chudasama, H. D. Nakum, D. K. Parmar, S. K. Gupta, P. K. Jha, Green Chem. Lett. Rev., 7, 278 (2014).
10) M. R. Maurya, M. Bisht, N. Chaudhary, F. Avecilla, U. Kumar, Polyhedron, 54, 180 (2013).
11) M. Moosavifar, S. Tangestaninejad, M. Moghadam, V. Mirkhani, J. Mol. Catal. A, 377, 92 (2013).
12) A. Okemoto, Y. Inoue, K. Ikeda, C. Tanaka, K. Taniya, Y. Ichihashi, S. Nishiyama, Chem. Lett., 43, 1734 (2014).
13) S. J. Kulkarni, C. N. Rohitha, N. Narender, A. Koeckritz, J. Porous Mater., 17, 321 (2010).
14) K. N. Bhagya, V. Gayathri, J. Porous Mater., 21, 197 (2014).
15) B. P. Nethravathi, K. R. Reddy, K. N. Mahedra, J. Porous Mater., 21, 285 (2014).
16) M. Madadi, R. Rahimi, Reac. Kinet. Mech. Cat., 107, 215 (2012).
17) A. Mobinikhaledi, M. Zendehdel, P. Safari, Reac. Kinet. Mech. Cat., 110, 497 (2013).
18) Z. Li, S. Wu, Y. Ma, H. Liu, J. Hu, L. Liu, Q. Huo, J. Guan, Q. Kan, Transition Met. Chem., 38, 243 (2013).
19) M. Ghorbanloo, S. Rahmani, H. Yahiro, Transition Met. Chem., 38, 725 (2013).
20) A. Nezamzadeh-Ejhieh, E. Shahriari, J. Ind. Eng. Chem., 20, 2719 (2014).
21) K. Mori, M. Kawashima, K. Kagohara, H. Yamashita, J. Phys. Chem. C, 112, 19449 (2008).
22) 森 浩亮,山下弘巳,ゼオライト,26, 131 (2009).
23) M. Martis, K. Mori, H. Yamashita, Dalton Trans., 43, 1132 (2014).
24) S. Rayati, E. Bohloulbandi, S. Zakavi, Inorg. Chem. Commun., 54, 38 (2015).
25) N. H. Khan, M. B. Ansari, E. A. Prasetyanto, H. Jin, S. Park, Tetrahedoron: Asymmetry, 22, 117 (2011).
26) H. Naeimi, M. Moradian, Appl. Catal. A, 467, 400 (2013).
27) H. Mei, J. Hu, S. Xiao, Y. Lei, G. Li, Appl. Catal. A, 475, 40 (2014).
28) R. Zhang, L. Liu, Y. Li, W. Wang, R. Li, Int. J. Electrochem. Sci., 10, 2355 (2015).
29) M. R. Avei, M. Jafarian, S. Etezadi, F. Gobal, M. Khakali, S. Rayati, M. G. Mahjani, Talanta, 108, 19 (2013).
30) R. Zhang, W. Zhang, L. Gao, J. Zhang, P. Li, W. Wang, R. Li, Appl. Catal. A, 466, 264 (2013).
31) S. Yamaguchi, T. Fukura, C. Fujita, H. Yahiro, Chem. Lett., 41, 713 (2012).
32) S. Yamaguchi, T. Fukura, K. Takiguchi, C. Fujita, M. Nishibori, Y. Teraoka, H. Yahiro, Catal. Today, 242, 261 (2015).
33) 山口修平,八尋秀典,化学工業,64, 22 (2013).
34) 山口修平,八尋秀典,触媒の設計・反応制御事例集,p. 224, 技術情報協会 (2013).
35) K. J. Balkus, Jr., A. G. Gabrielov, S. L. Bell, F. Bedioui, L. Roué, J. Devynck, Inorg. Chem., 33, 67 (1994).
36) I. L. V. Rosa, C. M. C. P. Manso, O. A. Serra, Y. Iamamoto, J. Mol. Catal. A, 160, 199 (2000).
37) H. Yahiro, K. Kimoto, H. Yamaura, K. Komaguchi, A. Lund, Chem. Phys. Lett., 415, 126 (2005).
38) M. Costas, M. P. Mehn, M. P. Jensen, L. Que, Jr., Chem. Rev., 104, 939 (2004).
39) B. Plietker ed., Iron Catalysis in Organic Chemistry: Reactions and Applications, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim (2008).
40) 山口修平,八尋秀典,ケミカルエンジニヤリング,60, 410 (2015).
41) S. Yamaguchi, T. Ohnishi, Y. Miyake, H. Yahiro, Chem. Lett., in press (2015). DOI: 10.1246/Cl.150479
42) S. Yamaguchi, H. Masuda, Sci. Tech. Adv. Mater., 6, 34 (2005).
43) A. Wada, M. Harata, K. Hasegawa, K. Jitsukawa, H. Masuda, M. Mukai, T. Kitagawa, H. Eingaga, Angew. Chem. Int. Ed., 37, 798 (1998).
44) T. Fujii, A. Naito, S. Yamaguchi, A. Wada, Y. Funahashi, K. Jitsukawa, S. Nagatomo, T. Kitagawa, H. Masuda, Chem. Commun., 2700 (2003).
45) 山口修平,鈴木彰規,八尋秀典,触媒,B, 68 (2014).